

China’s medical device market represents a huge opportunity for British manufacturers. Hamish King, COO of China-focused CRO and regulatory consultancy Cisema, explains how to navigate red tape to get your products ready for approval in China
The size of the Chinese healthcare industry is second only to the US, reaching a value of RMB 7.82 trillion in 2019. British medical device manufacturers have traditionally taken a cautious approach to the China market due to IP concerns, competition risks and fear about language and cultural barriers. But the market opportunity is huge for those who strategically structure their approach, instead of treating the market like another non-core area and delegating all activities to their distributor.
This article introduces the key steps manufacturers should take to help their medical device get approved by the China regulator by being seen as safe and effective for China sales.

Step 1: Determine the approval pathway for your device for China
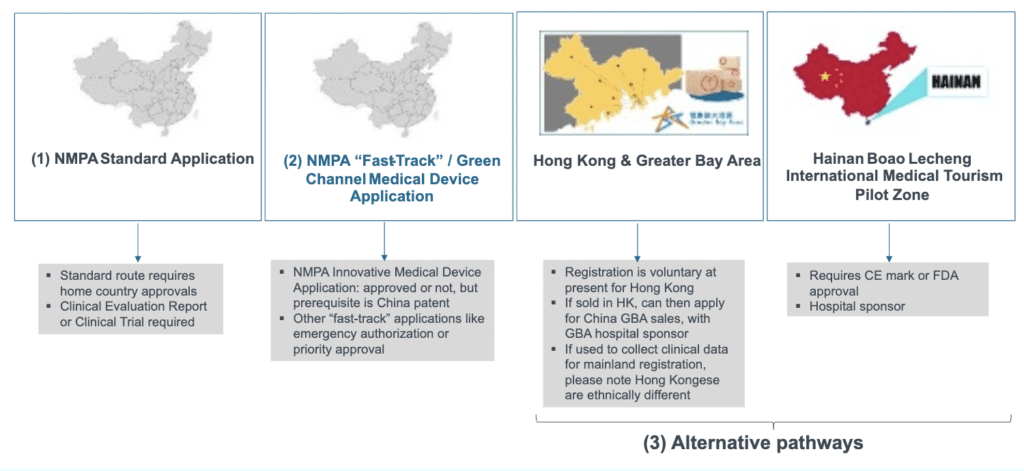
There are three broad pathways to get your medical device approved for sale into China:
- Standard approval process with the National Medical Products Administration (NMPA)
- Fast-track (also known as “green channel”) approval pathways
- Alternative pathways via sales to special areas within Greater China – the Greater Bay Area or Hainan Island Pilot Zone – that may enable easier registration via subsequent standard or fast-track approval processes.
The three main approval pathways to register a medical device in China
The vast majority of imported medical devices will seek to apply via the standard pathway to NMPA in Beijing. To do this, products should first have home country approval. For higher-risk devices (class II or III – see below for more information about classification), approval in China will probably require a detailed clinical evaluation report to be prepared or a clinical trial in China to be undertaken.
For “fast-track” pathways, only the newly introduced “Innovative” approval pathway allows devices without home country approvals to submit for China approval. Success in such applications requires a China patent, as well as evidence that the product is a first of its kind for application into China.
Other fast-track (also known as “green-channel”) approval pathways in China include priority review or emergency approval applications. Emergency approvals are reserved for special situations like the Covid-19 pandemic, and specific categories or types of devices will be announced by the NMPA as eligible for application. Once approved for emergency use, subsequent renewals will require the standard full documentation similar to standard applications.
The alternative pathways of the Hainan Pilot Zone and the Greater Bay Area are frameworks enabling companies with home country approvals to sell in limited circumstances. This can aid the collection of real-world data on China populations, which can reduce eventual China clinical trial requirements for subsequent NMPA standard or “fast-track” applications.
Step 2: Classify the device into the appropriate risk class and establish whether China clinical trials are required
Your medical device will need to be registered in your home market before you can apply for China marketing approval, subject to the exceptions explained above.
The risk classification in your home country jurisdiction will often be similar to China’s three risk classes, with class I being the lowest risk and class III being the highest risk (although not necessarily equivalent). According to the NMPA, the degree of risk is assessed based on “the intended purpose, structural characteristics, pattern of use, status of use as well as whether the device is body contacting.” China tends to be more conservative than other countries and so risk classification can be up-classified depending on the type of device.
There is an extensive risk classification catalogue which is sufficient to determine the appropriate class for most medical devices, but according to your author’s experience, in roughly 5% of cases it is necessary to apply directly to the regulator for a determination.

How to classify a medical device in China
If your product is a class I medical device then it will go through a simple filing process in China. If it is a class II or class III product, then it must go through a registration process which means there is a review of the product documentation by the NMPA prior to marketing approvals.
Class II and III device applications will need to be exempt from clinical trials, rely on predicate device data or be supported by clinical trial data (see Figure 3 for whether a clinical trial may be required).
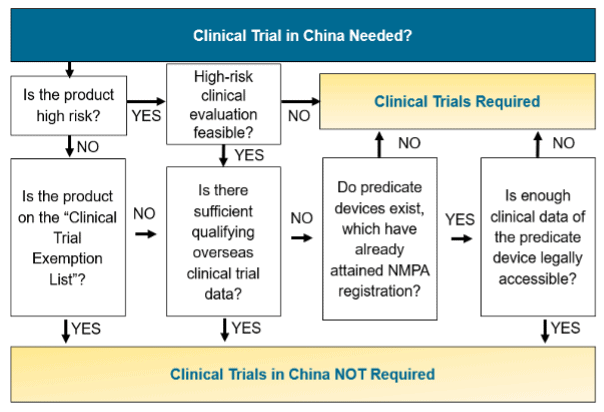
For companies that are unclear about whether China clinical trials are required, Cisema recommends preparing a feasibility study of the pathways and data available.
Step 3: Understanding the key steps for filing and registration
The filing process for class I is relatively straightforward and consists of a dossier preparation stage followed by submission to NMPA. This is represented in Figure 4 below. The total timeline is usually around 2-4 months.
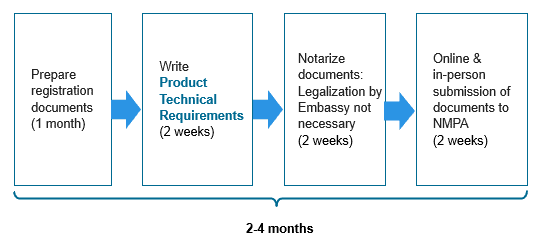
Filing process for class I submissions in China
For class II and III medical devices, the registration process is longer and more rigorous. First, there is the documentation stage, including drafting of the product technical requirements (PTR), which is a key technical protocol outlining the local testing specifications and relevant China standards. Then follows local testing of product samples in China – a mandatory requirement – following by clinical evaluation drafting or clinical trials, before submission of the dossier to NMPA for review and approval.
In certain cases, a class II or III product may be expressly exempt from clinical trials in China. In such cases, a simplified clinical evaluation report or even no CER at all will be required.
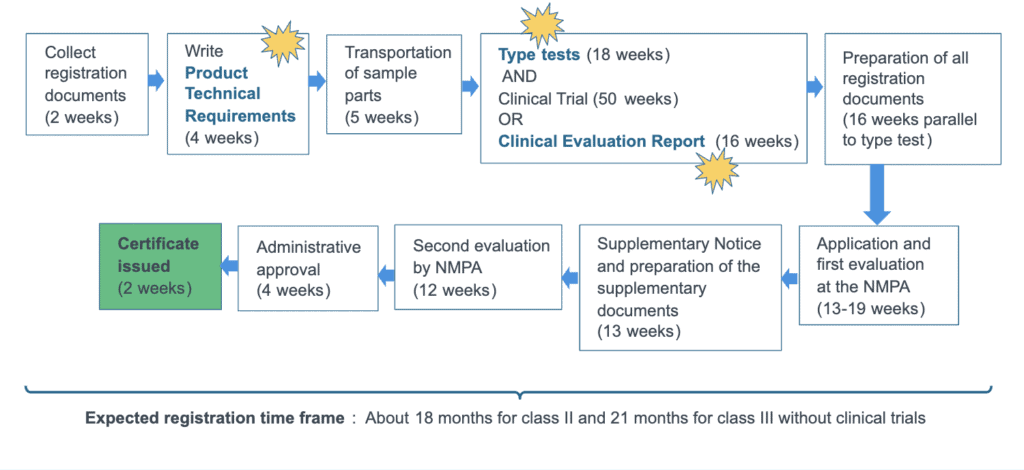
Timeline of China registration of class II & III medical devices
Step 4: Select your local China representative
In parallel with the registration process, manufacturers will need to carefully consider the identity of their local authorised representative – more accurately known as the NMPA Legal Agent. Like the EU and US, applicants must be represented by a local China entity for the purposes of submission and approval in China.
Importing manufacturers have three options: distributor, local subsidiary or service provider. The distributor option is often the default, but manufacturers should be cautious of potential IP risks (a lot of production information needs to be provided to the NMPA Legal Agent as part of the application process – and the dossier must be submitted by the agent) as well as for flexibility and regulatory expertise following marketing approvals. The NMPA Legal Agent will be involved in the first import of the product for each new distributor named, so flexibility will be limited for such a huge market if there is one master distributor who in effect needs to approve the others.
Also, NMPA has increasingly moved to a whole of lifetime perspective rather than point-of-time approvals. This means there will continue to be ongoing updates and requirements during the product’s lifetime even after marketing approvals are obtained. A typical list of your NMPA Legal Agent’s services should cover adverse events and recall management, labelling and regulatory mandatory updates, standards updates, periodic reporting and more.
Unless manufacturers want to make a substantial investment in their own subsidiary in China, a better option is to nominate an independent service provider who has deep regulatory expertise and fewer conflicts of interest.
In summary, UK manufacturers should think strategically about their China medical device registrations but not be swayed by hearsay or general fears. Registration of your device in China is realistic and achievable with the right approach and investment. Only then can China’s vast market potential be realised.
